








Serviços Personalizados
Artigo
 pdf em Português
pdf em Português Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailLinks relacionados
Compartilhar

 Permalink
PermalinkRevista da Associacao Paulista de Cirurgioes Dentistas
versão impressa ISSN 0004-5276
Rev. Assoc. Paul. Cir. Dent. vol.69 no.2 Sao Paulo Abr./Jun. 2015
Relato de caso clínico
Características bucais e craniofaciais da síndrome de Apert: relato de caso
Oral and craniofacial characteristics Apert syndrome: a case report
Alcides Ricardo GonçalvesI; Gabriela Cunha Bonini II; Tatiane Fernandes de NovaesIII; Tatiane MaregaIV; José Carlos Pettorossi ImparatoV
I Mestrando em Odontopediatria, especialista em Odontologia para Pacientes com Necessidades Especiais pelo CPO SL Mandic - Professor assistente do curso de especialização em Odontologia para Pacientes com Necessidades do CPO SL Mandic
II Doutora em Odontopediatria pela Faculdade de Odontologia da Universidade de São Paulo (Fousp) - Professora assistente de Odontopediatria da graduação do CPO SL Mandic e professora do programa de pós-graduação do CPO SL Mandic
III Pós-doutoranda em Ciências da Saúde pela Universidade Cruzeiro do Sul de São Paulo/SP, doutora em Odontopediatria pela Fousp - Professora convidada do programa de mestrado em Odontopediatria do CPO SL Mandic
IV Doutora em Educação Especial pela Universidade Federal de São Carlos (UFSCar) - Professora coordenadora do curso de especialização em Odontologia para Pacientes com Necessidades Especiais do CPO SL Mandic de Campinas/SP
V Livre-docência pela Fousp - Coordenador do programa de mestrado em Odontopediatria do CPO SL Mandic
RESUMO
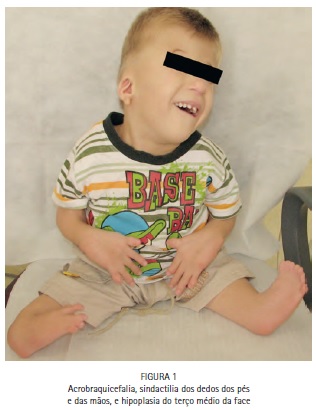
A síndrome de Apert, também conhecida como acrocefalossindactilia é uma rara condição genética caracterizada pela fusão prematura das suturas cranianas (craniossinostose), hipoplasia do terço médio da face e sindactilia das mãos e pés. O presente trabalho teve como objetivo demonstrar as principais características bucais e craniofaciais da síndrome de Apert, através de um relato de caso. Paciente do sexo masculino, 4 anos de idade, leucoderma, com diagnóstico médico de síndrome de Apert, foi encaminhado para a Clínica de Especialização em Odontologia para Pacientes com Necessidades Especiais do Centro de Pesquisas Odontológicas São Leopoldo Mandic, em Campinas, São Paulo, Brasil. No exame físico, notou-se a presença de sindactilia dos dedos dos pés e das mãos, alterações craniofaciais, hipoplasia da maxila, palato em forma de V invertido com pseudofenda na linha média, tumefações nos processos palatinos e ausência dos incisivos inferiores. Conclui-se que as características bucais e craniofaciais do paciente estão de acordo com as características relatadas na literatura para a síndrome, devendo o cirurgião-dentista estar apto a identificá-las.
Descritores: acrocefalossindactilia; pessoas com deficiência; odontopediatria
ABSTRACT
The Apert syndrome, also known as acrocephalosyndactyly, is a rare genetic condition characterized by the premature fusion of the cranial sutures (craniosynostosis), hypoplasy of the middle third of the face and syndactyly of the hands and feet. This study aimed to demonstrate the main oral and craniofacial characteristics of the Apert syndrome through a case report. A 4-year-old leucoderma male patient, with a diagnosis of Apert syndrome, was referred to the Clinic of Dentistry Specialization for Special Needs Patients at the Dental Research Center São Leopoldo Mandic, in Campinas, São Paulo, Brazil. On physical examination there have been noticed the presence of syndactyly of the toes and hands, craniofacial abnormalities, hypoplasy of the maxilla, inverted V shaped of the palate, with pseudocleft midline, swellings in the palatine processes and absence of the lower incisors. The results indicated that the oral craniofacial characteristics of the patient are in accordance with those reported in the literature for the Apert syndrome, and a dentist is required to identify them.
Descriptors: acrocephalosyndactylia; disabled persons; pediatric dentistry
RELEVÂNCIA CLÍNICA
A relevância clinica do presente trabalho é demonstrar as principais características bucais e craniofaciais da síndrome de Apert, através do relato de caso de uma criança acometida pela doença. É de extrema importância que o profissional Cirurgião-Dentista saiba reconhecer essas características no caso de pacientes com essa síndrome para que o tratamento adequado seja instituído.
INTRODUÇÃO
A síndrome de Apert, também conhecida como acrocefalossindactilia, recebeu esta denominação por ter sido descrita por Eugene Apert em 1906. É uma rara condição genética com padrão de transmissão autossômico dominante, caracterizada pela fusão prematura das suturas cranianas (craniossinostose), hipoplasia do terço médio da face e sindactilia das mãos e pés.1,2,3,4,5 Estima-se que a prevalência seja de aproximadamente 1:65.000 nascimentos2,6, com uma distribuição por sexo de 1:1.5
As malformações cranianas estão associadas a mutações ocorridas nos genes receptores do fator de crescimento de fibroblastos (FGFR), identificadas como agente etiológico de algumas síndromes com craniossinostose, incluindo a síndrome de Apert.7
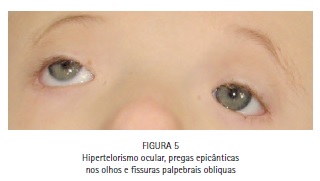
A fusão prematura de uma ou mais suturas cranianas acarreta deformação do crânio e aumento da pressão intracraniana8, sendo que os principais traços craniofaciais incluem: acrobraquicefalia (crânio em forma de torre), depressão dos ossos temporais, hipoplasia severa do terço médio da face, boca em forma de trapézio, ponte nasal achatada, hipertelorismo e proptose ocular, e fissuras palpebrais obliquas baixas.9
As manifestações bucais podem incluir: palato em forma de V invertido, maloclusão tipo classe III, tumefações ao longo da parte lateral do palato duro e pseudofenda palatina. Uma parcela dos pacientes podem apresentar fenda palatina e úvula bífida.2
Quanto à fusão dos dedos das mãos e pés, nome dado a essa condição de sindactilia, pode ocorrer apenas fusão dos tecidos moles como também fusão dos tecidos moles e ósseos.10 O diagnóstico pré-natal pode ser feito através da ultrassonografia e do estudo genético, este último viável apenas nos últimos anos.
O diagnóstico pós-natal, além do estudo genético, pode ser realizado através das características clínicas, radiografias, tomográficas e da ressonância magnética.4 O diagnóstico diferencial deve ser feito com outras síndromes que causam craniossinostose, como a síndrome de Pfeiffer, Crouzon e Saethre-Chotzen.11
O tratamento cirúrgico da síndrome de Apert é realizado para correção das deformações funcionais e estéticas do crânio e das sindactilias das mãos e pés, contribuindo para a melhora estética e funcional do paciente.12 O tratamento ortodôntico costuma ser necessário e é integrado ao tratamento cirúrgico para tratamento da craniossinostose.2
Este relato de caso teve como objetivo demonstrar as principais características bucais e craniofaciais da síndrome de Apert, sendo de extrema importância que o profissional Cirurgião- Dentista saiba reconhecer essas características no caso de pacientes com essa síndrome para que o tratamento adequado seja instituído.
RELATO DE CASO CLÍNICO
Paciente do sexo masculino, 4 anos de idade, leucoderma, com diagnóstico médico de síndrome de Apert, compareceu no Centro de Pesquisas Odontológicas São Leopoldo Mandic, de Campinas/SP, acompanhado de sua mãe, que tinha como queixa principal a ausência de alguns dentes decíduos, onde foi encaminhada para Clínica de Especialização em Odontologia para Pacientes com Necessidades Especiais.
Durante a anamnese colhida junto à sua mãe, que teve uma filha mais velha que não apresentava a síndrome, constatou- se que o paciente permanecia em tratamento médico no hospital de clínicas da Universidade Estadual de Campinas/ SP (Unicamp), não estava fazendo uso de nenhum medicamento, não havia sido submetido a procedimentos cirúrgicos estéticos e/ou funcionais até o momento e não falava, apenas emitia alguns sons.
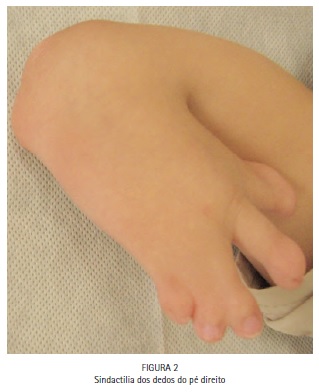
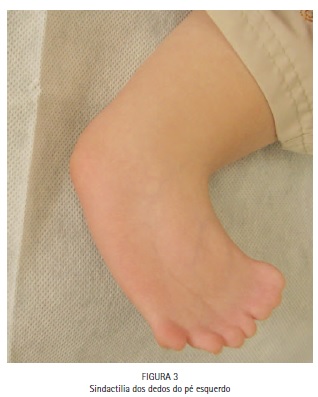
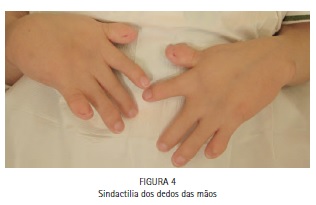
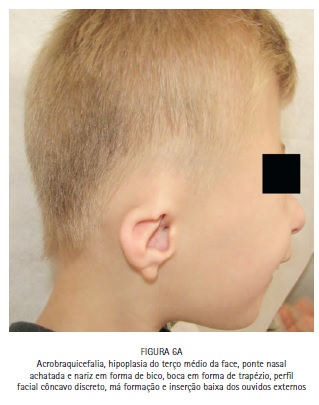
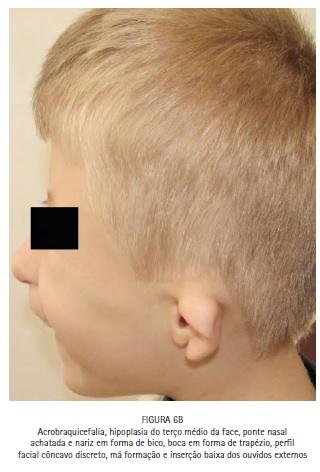
No exame físico, notou-se a presença de acrobraquicefalia (Figuras 1, 6A e 6B), sindactilia dos dedos dos pés (Figura 1, 2 e 3) e das mãos (Figura 1 e 4), hipoplasia do terço médio da face (Figuras 1, 6A e 6B), hipertelorismo ocular, pregas epicânticas nos olhos, fissuras palpebrais oblíquas (Figura 5), ponte nasal achatada e nariz em formato de bico, boca em forma de trapézio, perfil facial côncavo discreto, má formação e inserção baixa dos ouvidos externos. (Figuras 6A e 6B).
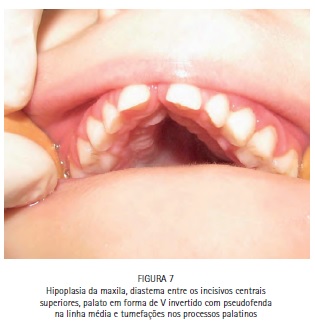
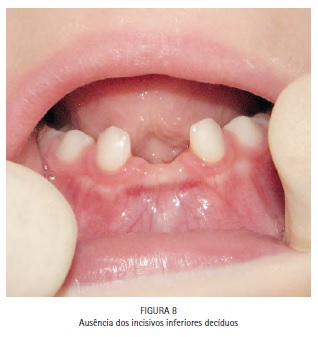
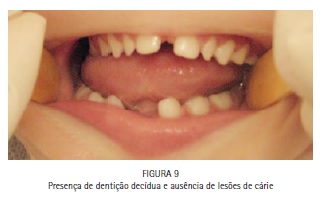
O exame físico intrabucal revelou hipoplasia da maxila, diastema entre os incisivos centrais superiores, palato em forma de V invertido com pseudofenda na linha média, tumefações nos processos palatinos (Figura 7), ausência dos incisivos inferiores decíduos (Figura 8), presença de dentição decídua e ausência de lesões de cárie (Figura 9).
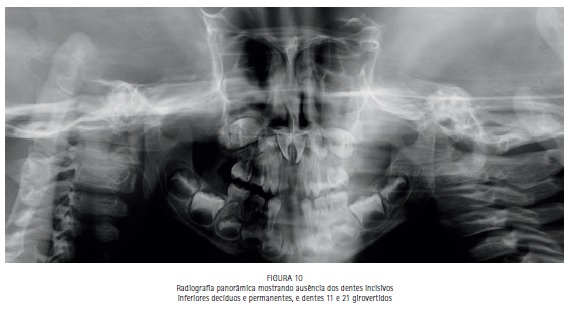
A radiografia panorâmica revelou ausência dos dentes incisi-
vos inferiores decíduos e permanentes, e dentes 11 e 21 girovertidos (Figura 10).
A conduta clínica requereu orientações quanto à técnica de higiene bucal, profilaxia e aplicação tópica de flúor. As consultas de rotina foram instituídas a cada seis meses, facilitando assim a manutenção da saúde bucal do paciente e o acompanhamento de possíveis agravos relacionados à síndrome.
DISCUSSÃO
As craniossinostoses são classificadas de acordo com a(s) craniana(s) sutura(s) afetada(s).13 A fusão prematura de uma ou mais suturas cranianas pode ocorrer como anormalidade primária, como parte de uma síndrome, ou estar associada a um conjunto de condições sistêmicas.8
O retardo mental é considerado usual para o paciente com síndrome de Apert e pode ocorrer devido às malformações cerebrais,
alta pressão intracraniana ou ambiente familiar.14,15,16
A ocorrência da síndrome de Apert pode estar associada à idade paterna avançada. O que pode explicar esta associação é que a replicação das células pré-meióticas durante a vida do homem resulta no acúmulo de mais mutações na linha germinativa de indivíduos mais velhos, assim elevando a frequência das mutações dos espermatozoides.17,18,19
A maior ocorrência das mutações genéticas acontece no cromossomo 10q (10q25-26), especialmente no lócus 10q26 do gene FGFR2 (receptor do fator de crescimento de fibroblastos 2).12 Entre as craniossinostoses sindrômicas causadas por mutação no FGFR2, a síndrome de Apert e a síndrome de Crouzon se destacam, representando os extremos do espectro de variabilidade clínica causada por ganho de função de mutações em FGFR2.6
O paciente do presente caso apresentou como características craniofaciais: acrobraquicefalia, hipoplasia do terço médio da face, hipertelorismo ocular, pregas epicânticas nos olhos, fissuras palpebrais oblíquas, ponte nasal achatada e nariz em formato de bico, boca em forma de trapézio, perfil facial côncavo discreto, má formação e inserção baixa dos ouvidos externos.1,20,21,22 Os achados bucais revelaram: hipoplasia da maxila, palato em forma de V invertido com pseudofenda na linha média, tumefações nos processos palatinos7,12,20,22,23,24,25,26 e ausência dos incisivos inferiores decíduos.27 A radiografia panorâmica revelou ausência dos dentes incisivos inferiores decíduos e seus sucessores permanentes27, e giroversão dos dentes 11 e 21.24 Esses achados corroboram com estudos prévios para o diagnóstico clínico da síndrome de Apert, mesmo ainda, no presente caso, o diagnóstico por meio de estudos genéticos não ter sido realizado.
A totalidade (100%) dos pacientes com síndrome de Apert apresentam acrobraquicefalia, hipoplasia do terço médio da face e sindactilia dos dedos30, sendo que essas características são marcantes no paciente relatado.
A ausência de alguns dentes decíduos e/ou permanentes podem ter uma frequência de até 44,4%, as tumefações nos processos palatinos podem ocorrer em até 88,8% e a erupção ectópica também pode estar presente em 33,3% dos casos.12 Essas alterações podem ser explicadas pela hipoplasia da maxila12 que, também, está presente no referido paciente.
Embora a hipoplasia da maxila resultar em uma significante maloclusão tipo classe III22,28, o paciente relatado não apresentava esta característica, sendo que o mesmo será acompanhado clinicamente sempre observando essa possível condição. A fenda palatina, presente em até 24% dos pacientes, e a úvula bífida, em até 75%31, também não foram características apresentadas pelo paciente.
O tratamento cirúrgico das craniossinostoses visa melhorar a estética e previnir alterações neurológicas.12
A craniostomia pode ser realizada durante o 1° ano de vida para tratar a craniossinostose. O avanço frontofacial e do terço médio da face podem ser feitos depois para corrigir a proptose e a hipoplasia do terço médio da face.4
O tratamento ortodôntico costuma ser necessário e é integrado ao tratamento cirúrgico.2 Não há um consenso quanto o melhor tempo para corrigir cirurgicamente a hipoplasia do terço médio da face, sugere-se que a cirurgia deve ocorrer de nove a doze anos de idade com tratamento ortodôntico dos 12 aos 21 anos.29
O tratamento deve ser realizado por uma equipe interdisciplinar, sendo que o planejamento cirúrgico ocorre em diferentes etapas: correção das alterações craniofaciais (descompressão cirúrgica para crescimento cerebral normal), avanço do terço médio da face (melhorar o fluxo aéreo-nasal) e cirurgia ortognática (correção funcional e estética). A cirurgia corretiva para a sindactilia dos dedos das mãos deve ser realizada na infância, enquanto que para os dedos dos pés a cirurgia corretiva está indicada se a habilidade de andar estiver prejudicada.31 Até o momento o referido paciente não havia sido submetido a procedimentos cirúrgicos estéticos e/ou funcionais.
O paciente será acompanhado ambulatorialmente na Clínica de Especialização em Odontologia para Pacientes com Necessidades Especiais do Centro de Pesquisas Odontontógicas São Leopoldo Mandic, de Campinas/SP, visando à manutenção da saúde bucal, o diagnóstico e o tratamento precoce de possíveis alterações inerentes à síndrome; assim como possíveis reabilitações orais no momento oportuno. A interdisciplinaridade será estabelecida junto à equipe médica do hospital de clínicas da Universidade Estadual de Campinas/SP (Unicamp), sempre visando condutas conjuntas para o bem-estar do paciente.
CONCLUSÃO
De acordo com o relato de caso apresentado, conclui-se que as principais características bucais e craniofaciais do paciente estão de acordo com as características relatadas na literatura para a síndrome, devendo o Cirurgião-Dentista estar apto a identificá-las.
APLICAÇÃO CLÍNICA
Esse relato de caso demonstra de maneira efetiva as principais características bucais e craniofaciais da síndrome de Apert, fazendo com que o Cirurgião-Dentista clínico geral ou especialista esteja apto a identificá-las e, desta maneira, instituir o tratamento ou direcionamento adequado a seu paciente.
AGRADECIMENTOS
Ao programa de Mestrado em Odontopediatria e ao Curso Especialização em Odontologia para Pacientes com Necessidades Especiais do Centro de Pesquisas Odontológicas São Leopoldo Mandic, de Campinas/SP.
REFERÊNCIAS
1. Gorlin RJ, Cohen MM Jr, Levin LS. Syndromes with craniosynostosis: general aspects and well-known syndromes. 4nd ed. New York: Oxford University Press; 2001. [ Links ]
2. Neville BW, Damm DD, Allen CM, Bouquot JE. Patologia oral e maxillofacial. 3a ed. Rio de Janeiro: Elsevier; 2009.
3. Surman TL, Logan RM, Townsend GC, Anderson PJ. Oral features in Apert syndrome: a histological investigation. Orthod Craniofac Res. 2010 Feb;13(1):61-67.
4. Bhatia PV, Patel PS, Jani YV, Soni NC. Apert's syndrome: report of a rare case. Journal of Oral and Maxillofacial Pathology. 2013 May;17(2):294-7.
5. Pi G, Zúñiga A, Cervera J, Ortiz M.. Diagnóstico prenatal de síndrome de Apert por mutación de novo en gen FGFR2. An Pediatr. 2014;80(3):e104-e5.
6. Yeh E, Fanganiello RD, Sunaga DY, Zhou X, Holmes G, Rocha KM, et al. Novel molecular pathways elicited by mutant FGFR2 may account for brain abnormalities in Apert Syndrome. Plos One. 2013 Apr 4;8(4):1-7.
7. Şoancӑ A, Dudea D, Gocan H, Roman A, Culic B. Oral manifestations in Apert syndrome: case presentation and a brief review of the literature. Romanian Journal of Morphology and Embryology. 2010;51(3):581-4.
8. Kouga T, Tanoue K, Matsui K. Airway statuses and nasopharyngeal airway use for airway obstruction in syndromic craniosynostosis. The Journal of Craniofacial Surgery. 2014 May;25(3):762-5.
9. Ileri Z, Goyenc YB. Apert syndrome: a case report. European Journal of Dentistry. 2012 Jan;6(1):110-3.
10. Zhou J, Chen Y, Cao K, Zou Y, Zhou H, Hu F, et al. Functional classification and mutation analysis of a synpolydactyly kindred. Experimental and Therapeutic Medicine. 2014 Nov;8(5):1569-1574.
11. Múfalo PS, Kaizer RO, Dalben GS, de Ameida AL. Comparasion of periodontal parameters in individuals with syndromic craniosynostosis. J Appl Oral Sci. 2009 Jan-Feb;17(1):13-20.
12. Dalben GS, Neves LT, Gomide MR. Oral findings in patients with Apert syndrome. J Appl Oral Sci. 2006 Dec;14(6):465-9.
13. Trad CS, Rosique RG. Craniossinostose primárias: ensaio iconográfico. Radiol Bras. 2005;38(5):377-380.
14. Yacubian-Fernandes A, Palhares A, Giglio A, Gabarra RC, Zanini S, Portela L, et al. Apert Syndrome: factors involved in the cognitive development. Arq Neuropsiquatr. 2005;63(4):963-8.
15. de Jong T, Rijken BFM, Lequin MH, van Veelen MLC, Mathijssen IMJ. Brain and ventricular volume in patients with syndromic and complex craniosynostosis. Childs Nerv Syst. 2012 Jan;28(1):137-140.
16. Liu J, Kwon TG, Nam HK, Hatch NE. Craniosynostosis-associated Fgfr2 (C342Y) mutant bone marrow stromal cells exhibit cell autonomous abnormalities in osteoblast differentiation and bone formation. BioMed Research International. 2013 Mar.
17. Lajeunie E, Heuertz S, El Ghouzzi V, Martinovic J, Renier D, Le Merrer M, et al. Mutation screening in patients with syndromic craniosynostoses indicates that a limited number of recurrent FGFR2 mutations accounts for severe forms of Pfeiffer syndrome. European Journal of Human Genetics. 2006 Mar;14(3):289-298.
18. Yoo SR, Qin J, Glaser RL, Jabs EW, Wexler NS, Sokol R, et al. The ups and downs of mutation frequencies during aging can account for the Apert syndrome paternal age effect. PLoS Gentics. 2009 Jul;5(7).
19. Goriely A, Wilkie AO. Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. The American Journal of Human Genetics. 2012 Feb 10;90(2):175-200.
20. Tosun G, Sener Y. Apert syndrome with glucose‐6‐phosphate dehydrogenase deficiency: a case report. International Journal of Paediatric Dentistry. 2006 May;16(3):218-221.
21. Martelli H Jr, Paranaíba LM, de Miranda RT, Orsi J Jr, Coletta RD. Apert syndrome: report of a case with emphasis on craniofacial and genetic features. Pediatric Dentistry. 2008 Nov-Dec;30(6):464-8.
22. Vadiati Saberi B, Shakoorpour A. Apert syndrome: report of a case with emphasis on oral manifestations. Journal of Dentistry Tehran University of Medical Sciences. 2011;8(2):90-5.
23. Letra A, de Almeida AL, Kaizer R, Esper LA, Sgarbosa S, Granjeiro JM. Intraoral features of Apert's syndrome. Oral Surgery Oral Medicine Oral Pathology Oral Radiology and Endodontics. 2007 May;103(5):e38-41.
24. Carneiro GVS, Farias JG, Santos FAP, Lamberti PL. Síndrome de Apert: revisão de literatura e relato de um caso clínico. Rev Bras Otorrinolaringol. 2008;74(4):640.
25. Kahnberg KE, Hagberg C. Orthognathic surgery in patients with craniofacial syndrome. I. A 5-year overview of combined orthodontic and surgical correction. J Plast Surg Hand Surg. 2010 Dec;44(6): 282-8.
26. Costa FW, Rodrigues RR, Batista AC, Ribeiro TR, Pereira KM. Multiple radiopaque mandibular lesions in a patient with Apert syndrome. J Endod. 2012 Dec;38(12):1639-43.
27. Reitsma JH, Elmi P, Ongkosuwito EM, Buschang PH, Prahl-Andersen B. A longitudinal study of dental arch morphology in children with the syndrome of Crouzon or Apert. Eur J Oral Sci. 2013 Aug;121(4):319-327.
28. Derderian C, Seaward J. Syndromic craniosynostosis. Semin Plast Surg. 2012 May;26: 64-75.
29. Oberoi S, Hoffman WY, Vargervik K. Craniofacial team management in Apert syndrome. American Journal of Orthodontics and Dentofacial Orthopedics. 2012 Apr;141(4 Suppl):S82-7.
30. Carrera IA, Martínez-Frías ML, Pérez JJM, Grisolía LP, Rodríguez AC, Conde CN, et al. Síndrome de Apert: análisis clínico-epidemiológico de una serie consecutiva de caso en España. Medicina Fetal y Neonatologia. 1999;51(6):667-676.
31. Castro-Silva II, Nascimento LA, Coutinho LACR, Costa DOP. Criança com síndrome de Apert: diagnóstico clínico-radiográfico, manifestações orofaciais e qualidade de vida. Rev Odontol Bras Central. 2014;23(66):151-4.
 Autor para correspondência:
Autor para correspondência:
Alcides Ricardo Gonçalves
Rua Antonio Pedro, 97
Centro – Pedreira/SP
13920-000
Brasil
e-mail: ricardo.pacientesespeciais@gmail.com
Recebido: fev/2015
Aceito: abr/2015