








Serviços Personalizados
Artigo
 pdf em Português
pdf em Português Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailLinks relacionados
Compartilhar

 Permalink
PermalinkArquivos em Odontologia
versão impressa ISSN 1516-0939
Arq. Odontol. vol.48 no.1 Belo Horizonte Jan./Mar. 2012
RELATO DE CASO
Displasia Ectodérmica: relato de caso
Ectodermal Dysplasia: case report
Christiane Santos FerreiraI; Rúbia Alves Marques Hissa FerreiraI; Maria Luíza M. F. FernandesI; Kelly Moreira Grillo Ribeiro BrancoI; Rodrigo Rezende ArantesII; Letícia Lima LeãoII
ICurso de Odontologia, Centro Universitário Newton Paiva, Belo Horizonte, MG, Brasil
IIServiço Especial de Genética, Hospital das Clínicas, Universidade Federal de Minas Gerais (UFMG), Belo Horizonte, MG, Brasil
Contato: chrissantosferreira@gmail.com, rubiahissa@gmail.com, mattaml@terra.com.br, kellybranco@iecinternet.com.br, rrarantes@hotmail.com, leticia_leao@uol.com.br
RESUMO
A displasia ectodérmica (DE) compreende um grupo grande e heterogêneo de doenças hereditárias que se caracteriza por apresentar manifestações clínicas relacionadas às anomalias das estruturas de origem ectodérmica, principalmente nos cabelos, unhas, dentes e pele. Este trabalho descreve o caso clínico de um paciente do sexo masculino de 11 anos de idade, que compareceu à clínica odontológica de uma instituição de ensino superior de Belo Horizonte MG. A criança apresentava-se com características da displasia ectodérmica, na forma hipoidrótica, e com história de diversos indivíduos afetados na família materna. Fez-se, ainda, revisão da literatura e discussão da etiologia e tratamento para o caso descrito.
Descritores: Displasia ectodérmica. Anodontia. Adolescente.
ABSTRACT
Ectodermal dysplasia includes a large and heterogeneous group of hereditary diseases characterized by clinical manifestations related to alterations in ectodermal structures, mainly hair, nails, teeth, and skin. The present study describes a clinical case of an eleven-year-old male patient that sought out dental services at a dentistry clinic in a higher education institution in Belo Horizonte, Brazil. The patient presented characteristics of ectodermic dysplasia, in hypohidrotic form, and a medical record of a number of members of the maternal family who had also been affected. A literature review and a discussion concerning the etiology and treatment for the described case was also carried out.
Uniterms: Ectodermal dysplasia. Anodontia. Adolescent.
INTRODUÇÃO
A displasia ectodérmica (DE) constitui um complexo grupo de doenças hereditárias. Existem mais de 150 síndromes, clinicamente distintas, em que a DE está presente1-2. A prevalência na população varia de 1:10.000 a 1:100.000 nascimentos e a proporção entre os sexos é de cinco homens para uma mulher3-4.
É de extrema importância que o cirurgiãodentista saiba identificar as principais características da DE, porquanto não são raros os casos de pacientes portadores dessa síndrome que chegam à clínica odontológica. Mesmo considerando que os indivíduos acometidos apresentem fácies característica, as manifestações clínicas e físicas são variáveis e estão relacionadas à heterogeneidade genética.
O diagnóstico correto e precoce é primordial para restabelecer as funções estética, mastigatória, fonética e psicológica desses pacientes, reintegrando-os ao convívio social.
REVISÃO DE LITERATURA
As DE constituem um grupo de doenças genéticas nas quais ocorrem diversas anomalias nas estruturas derivadas do folheto ectodérmico1,3,6-13. As alterações mais comumente encontradas decorrem de defeitos da epiderme e seus anexos: cabelo, dentes, unhas, glândulas sudoríparas e sebáceas1,4-8,12-17. Entre as anomalias faciais encontradas citam-se: nariz em sela, lábios protrusos, orelhas mal formadas com implantação oblíqua, regiões supraciliares salientes. Ocorre perda de dimensão vertical de oclusão devidoà ausência de elementos dentários, associada à presença de discretas fissuras ao redor da boca e olhos, o que determina uma aparência de senilidade. A redução ou ausência da lâmina dental leva os incisivos e caninos a apresentarem forma conoide. Geralmente acometem as duas dentições, ocorrendo hipoplasia de esmalte e podendo, ou não, haver anodontia4,6-8,18.
As DE podem ser classificadas de acordo com a produção de suor em duas formas: hidrótica, em que há produção de suor, e hipoidrótica, em que a produção de suor é reduzida ou ausente19. Na forma hidrótica, ou síndrome de Clouston, as principais alterações encontradas são: unhas distróficas, pelos escassos e anomalias dentárias, não havendo, entretanto, comprometimento das glândulas sudoríparas e sebáceas6,7. O padrão de herança genética é do tipo autossômico dominante e, por isso, observa-se a mesma frequência em ambos os sexo2,19. Na displasia ectodérmica hipoidrótica (DEH), que é a forma mais comum, o padrão de herança em 95% dos casos é recessivo ligado ao cromossomo X. Os 5% restantes apresentam etiologia autossômica dominante e autossômica recessiva4,11-13,9-21.
Nos casos com herança recessiva ligada ao cromossomo X, os homens apresentam expressão mais completa da síndrome. As mulheres heterozigotas, portadoras do alelo recessivo, se apresentam clinicamente normais ou levemente afetadas, fenômeno explicado pela inativação aleatória de um dos cromossomos X no início do período embrionário (hipótese de Lyon)8,16,18.
Os sinais cardinais da DEH são: hipotricose (diminuição de cabelos), hipoidrose (diminuição na sudorese) e hipodontia (ausência congênita de alguns dentes). Algumas alterações estão presentes ao nascimento, enquanto outras só tornam-se evidentes durante a infância19.
Como consequência da alteração nas glândulas sudoríparas na DEH, observa-se a diminuição do suor, provocando elevação da temperatura corporal, ocorrência de crises de hipertermia e convulsões febris. A pele, geralmente hiperpigmentada, torna-se fina e desidratada, muitas vezes descamativa, podendo ser sede de dermatites atópicas, xerodermia e placas de liquenificação1.
As malformações dentárias encontradas com mais frequência são anodontia completa ou parcial da dentição decídua e/ou permanente, podendo causar a ausência ou deficiência alveolar. As anomalias mais frequentes ocorrem nos incisivos e caninos. O molar é o segundo dente mais afetado e apresenta taurodontismo8,21.
Uma complicação comumente encontrada em portadores da displasia ectodérmica é a rinite atrófica que, geralmente, acomete crianças na segunda infância ou adolescentes; a qual se caracteriza por uma inflamação nasal crônica, com produção excessiva de secreções fétidas devido a movimentos deficientes dos cílios nasais1.
A displasia ectodérmica pode ser confundida, ou fazer parte de outras síndromes como a E.E. C (Ectrocactilia, displasia ectodérmica e fenda palatina) e a síndrome trico-rino-falangeal que, apesar do aspecto facial semelhante, apresenta outras características como falanges distais curtas nas mãos, deformidade das articulações Inter falangeanas e epífises com forma cônica12,23.
Por meio de estudos genéticos, foi mostrado que cerca de 94% dos casos da doença ocorrem por mutação no gene EDA, situado no braço longo do cromossomo X (Xq12-q13.1)1,8-9,21-24. Os genes EDAR e EDARADD são associados com as formas autossômica dominante e autossômica recessiva respectivamente. Mutações nesses genes são responsáveis por 5% dos casos de DEH19.
A patogênese molecular da DEH ainda é pouco compreendida. O gene EDA determina a produção da ectodisplasina A, uma proteína necessária ao desenvolvimento de algumas estruturas derivadas do ectoderma, como cabelos, dentes e glândulas sudoríparas. Essa proteína parece ser importante em diversas vias que envolvem interações ectodermamesoderma, durante a embriogênese19.
Embora os indivíduos afetados, independentemente do tipo de herança, possam apresentar características clínicas semelhantes, a identificação da forma de transmissão é importante para que possa ser feito o aconselhamento genético à família. O exame completo dos familiares dos pacientes com DE e a identificação dos indivíduos que apresentam as formas parciais da doença na família são necessários para se esclarecer a transmissão genética naquele grupo6.
No manejo dos pacientes com DE deve-se abordar as alterações relacionadas aos sinais cardinais da doença e os objetivos principais são: prevenir a hipertermia, estabelecer uma boa função oral e melhorar o desenvolvimento psicossocial25.
A hipotricose deve ser tratada pelo dermatologista com fórmulas e técnicas cirúrgicas. Em alguns casos pode ser sugerido o uso de perucas. Com relação à hipoidrose, os principais cuidados são direcionados para diminuir as crises de hipertermia e suas complicações, são indicados banhos frios, roupas leves, pouca atividade física e a procura de climas mais amenos25. O prognóstico desta doença é de sobrevida até a idade adulta, desde que haja controle da temperatura corporal, porém dados da literatura mostram que 30% dos meninos morrem durante os primeiros dois anos de vida por hiperpirexia e infecções respiratórias20.
Para o acompanhamento dos indivíduos acometidos é necessário ter uma equipe multidisciplinar composta por médicos (geneticista, pediatra, otorrinolaringologista e dermatologista), cirurgiões dentistas (odontopediatra, protesista e implantodontista), fonoaudiólogos e psicólogos4-6,8.
O tratamento odontológico consiste em movimentações ortodônticas, confecção de facetas estéticas, próteses parciais ou totais, reconstrução estética com compósitos e implantes osseointegrados. A reabilitação dentária, além de melhorar as funções mastigatórias e fonéticas, resgata a autoestima e possibilita melhor integração social desses indivíduos4-6,10.
Sugere-se que deva ser feita a colocação de implantes tão logo a criança faça três anos de idade, possibilitando a finalização do tratamento antes da puberdade, e um desenvolvimento funcional e psicossocial ótimo. Porém, considera-se que a colocação de implantes ósseo integrados na fase de crescimento pode acarretar trauma aos germes dos dentes permanentes, desordens na erupção dos dentes e restrições multidimensionais em relação crescimento craniofacial10.
Após o diagnóstico da DE é essencial orientar os pais ou responsáveis em relação aos problemas relacionados à diminuição da sudorese e necessidade de monitorar o calor. Problemas otológicos, conjuntivais, hipofisários, respiratórios e gastrointestinais precisam ser investigados pelos profissionais competentes. Ao cirurgião-dentista cabe tratar as anormalidades dentárias, reabilitar o paciente, além de controlar a deficiência da saliva e suas conseqüências6.
CASO CLÍNICO
Paciente do sexo masculino, onze anos de idade, foi encaminhado pelo Núcleo de Apoio à Família (NAF) à clínica Odontológica do Centro Universitário Newton Paiva, em Belo Horizonte MG. A queixa principal, apresentada pela mãe era: "Ele precisa de tratamento mais específico porque não tem alguns dentes", esta relatou ainda, que a criança até dezoito meses de idade, não havia tido erupção dentária, levando-a a procurar ajuda médica. Foi orientada a buscar avaliação odontológica, sendo esta realizada quando a criança completou nove anos de idade. A temperatura corpórea aumentava mesmo durante o repouso. A mãe, quando questionada, relatou que ele não transpirava. De acordo com a anamnese, o paciente era o primeiro filho de pais não consanguíneos, pai saudável e mãe apresentando ausência dos incisivos laterais superiores e dos terceiros molares superiores e inferior do lado esquerdo, sendo classificado como hipodontia. O mesmo paciente possui um irmão com nove anos de idade e uma meio-irmã paterna de quinze anos, ambos sem doença genética. A mãe nega o uso de teratógenos durante o período gestacional. Segundo relato, na família materna há indivíduos com hipodontia em quatro gerações consecutivas, a partir do bisavô.
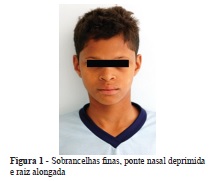
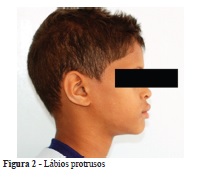
Foi realizada avaliação clínica do paciente no Serviço Especial de Genética do Hospital das Clínicas da UFMG (HC/UFMG), onde os seguintes sinais foram identificados: pele ressecada e fina, sobrancelhas finas, cílios rarefeitos, ponte nasal deprimida e raiz alongada, lábios grossos, oligodontia, caninos conoides, hipotricose corporal, hipoidrose e desenvolvimento neuropsicomotor adequado para a idade (Figuras 1 e 2).
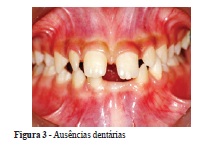
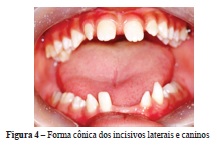
Ao exame clínico intrabucal realizado na clínica odontológica do Centro Universitário Newton Paiva, constatou-se a ausência dos dentes 18, 17, 28, 27, 38, 37, 31, 41 e 48. Além disso, os caninos e incisivos laterais superiores e inferiores apresentavam forma conoide; o dente 42 encontrava-se mesializado devido à ausência do dente 41, havia mordida cruzada dentária do dente 26. Constatou-se, ainda, má higienização bucal (Figuras 3 e 4).
Foi solicitada radiografia panorâmica para avaliação dos germes dentários dos permanentes. Na qual, verificou-se a ausência dos seguintes germes dentários: 18, 17, 28, 27, 38, 37, 31, 41 e 48, classificando, assim, o paciente como portador de oligodontia. Entretanto, maxila e mandíbula apresentavam desenvolvimento normal.
O tratamento odontológico proposto incluiu a adequação do paciente, por meio de ações educativas em saúde bucal, com intuito de elevar a autoestima do paciente, melhorar a higienização bucal, participação e interesse no tratamento. A seguir, realizou-se a colagem de um botão na vestibular do dente 36 e na palatina do dente 26, para colocação de um elástico 1/8, com o objetivo de descruzar a mordida, além da confecção de prótese parcial removível para manutenção dos espaços dentários, até o paciente completar a fase de crescimento e desenvolvimento craniofacial, para posterior planejamento protético definitivo, com confecção de próteses fixas ou implante ósseo integrado.
Com a colocação da prótese parcial removível na região anterior, restabeleceu-se a estética e a fonética e foi eliminado o hábito de interposição lingual. Também se corrigiu o posicionamento do dente 42, este foi vestibularizado por meio de uma mola digital (Figuras 5 e 6).
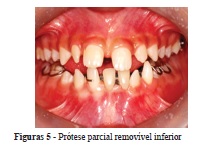
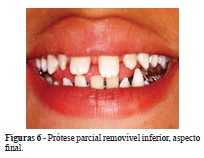
O paciente tem sido avaliado a cada três meses para acompanhamento do descruzamento do dente 26, da dentição irrompida e não irrompida e para possíveis ajustes na prótese parcial removível, pois não é necessária sua troca nessa fase, por não haver alterações dimensionais no arco dentário inferior.
Na última consulta, a mãe do paciente relatou que ele estava desestimulado e negligente em alguns aspectos do tratamento proposto, como exemplo, deixando de usar o elástico, uma vez que, este deveria ser mantido, no período de um mês, durante as vinte e quatro horas do dia. Assim, o acompanhamento passou a ser mensal. Concluído o descruzamento dentário completo, o acompanhamento do paciente passará a ser semestral, até que se possa realizar o tratamento protético definitivo. Além disso, o paciente será acompanhado periodicamente no Serviço Especial de Genética do HC/UFMG.
DISCUSSÃO
O paciente do caso clínico descrito apresenta características compatíveis com o diagnóstico de displasia ectodérmica hipoidrótica. Em relação à etiologia, considerando-se que existe clara transmissão vertical no heredograma, expressão variável entre os afetados de ambos os sexos e que as alterações da pele e cabelos são discretas, provavelmente seja da forma autossômica dominante. Porém, a forma recessiva ligada ao X não pode ser descartada, pois não há relato de transmissão entre pessoas do sexo masculino. O acompanhamento desta alteração envolve uma equipe multidisciplinar composta por médicos (geneticista, dermatologista e otorrinolaringologista), cirurgiões-dentistas (odontopediatra, protesista e implantodontista), fonoaudiólogos e psicólogos, no intuito de melhorar as condições físicas, estéticas e funcionais; também, restabelecer o estado emocional do paciente e permitir a reintegração ao convívio social, estando de acordo com a literatura consultada4-6,8.
O tratamento deverá ser realizado de forma sintomática para prevenir ou amenizar os sintomas. Nesse caso, o paciente foi orientado a usar roupas leves, evitar atividades físicas extenuantes, tomar banhos frios e ingerir líquidos frequentemente para manter a temperatura corpórea adequada. O mesmo, ainda, foi encaminhado ao otorrinolaringologista para que fossem identificadas as manifestações correlacionadas que possam estar associadas à displasia ectodérmica hipoidrótica, devido às alterações secretórias das mucosas1-6.
O tratamento proposto ao paciente foi àconfecção de prótese parcial removível inferior com acompanhamento periódico, a fim de não acarretar dano ao crescimento craniofacial do mesmo. De acordo com a literatura consultada, esse tratamento seria o de melhor escolha, por se tratar de um paciente em fase de crescimento4-6,8,11.
O implante ósseo integrado seria outra opção de tratamento. Entretanto, existem divergências na literatura quanto à idade na qual deve ser realizado, sendo sugerido o implante ósseo integrado somente quando o paciente já tiver atingido a idade adequada4-6,8,11.
A reabilitação mandibular com prótese removível fabricada em resina acrílica torna-se difícil devido ao crescimento mandibular e altura deficiente do processo alveolar, o que resulta em insuficiências funcionais recorrentes da prótese inferior. Os implantes devem ser colocados quando a criança completar três anos de idade, finalizando o tratamento antes da puberdade, para proporcionar um desenvolvimento funcional e psicossocial ótimo. Contudo, descrevemse alguns efeitos potencialmente desfavoráveis na utilização de implantes ósseo integrados nestes pacientes em fase de crescimento, tais como, trauma aos germes dos dentes permanentes, desordens na erupção e restrições multidimensionais em relação ao crescimento craniofacial10. Assim, alguns autores consideraram mais prudente a indicação de implante ósseo integrado após o término da fase de crescimento4-6,8,11.
O paciente, neste estudo, foi encaminhado, também, ao dermatologista, por apresentar pele fina e quebradiça. Foi orientado ainda, a fazer um acompanhamento psicológico a fim de melhorar a adaptação social.
A avaliação realizada no Serviço de Genética do HC/UFMG por geneticistas teve o objetivo de confirmar o diagnóstico. O aconselhamento genético do paciente será realizado em idade apropriada, por razões éticas.
O prognóstico do paciente relatado é bom, desde que os cuidados com a manutenção preventiva da saúde física e bucal sejam mantidos.
CONSIDERAÇÕES FINAIS
A DE é uma doença hereditária que apresenta alterações em estruturas originárias do ectoderma, principalmente pelos, glândulas e dentes, mostrando características mais marcantes e predominantes no sexo masculino, quando a herança é ligada ao cromossomo X, enquanto na forma autossômica dominante o fenótipo se apresenta de forma mais leve.
O cirurgião-dentista deve estar apto a identificar e diagnosticar as DE, para que seja realizado correto tratamento. Para isto, é preciso que se faça uma anamnese minuciosa e que se conheçam as anomalias próprias desse grupo de doenças.
O tratamento odontológico na infânciaé multidisciplinar envolvendo as especialidades como a ortodontia, a prótese e a dentística, associado às especialidades médicas (otorrinolaringologistas, geneticistas e dermatologistas), bem como acompanhamento de fonoaudiólogo e psicólogo. A reabilitação com implante ósseo integrado e prótese fixa é a etapa final para o término do tratamento do caso clínico iniciado na infância.
O controle periódico deve ser estipulado para acompanhamento e reforços nas instruções de manutenção da saúde bucal.
REFERÊNCIAS
1. Koerner HN, Bettega S, Mocellin M. Rinite atrófica: relato de caso associado a displasia ectodérmica. Arq Int Otorrinolaringol. 2006; 10:1-6. [ Links ]
2. McKusick VA. Mendelian inheritance in man and its online version, OMIM. Am J Hum Genet. 2007; 80:588–604.
3. Corrêa MS, Ulson RC, Rodrigues CR, Azevedo AM. Displasia ectodérmica hereditária: revista da literatura com relato de um caso clínico. Rev Paul Odontol. 1997; 19:30-4. [ Links ]
4. Shigli A, Reddy RP, Hugar SM, Deshpande D. Hypohidrotic ectodermal dysplasia: A unique approach to esthetic and prosthetic management. J Indian Soc Pedod Prev Dent. 2005; 23:31-4. [ Links ]
5. Assumpção M, Modesto A, Ruschel H, Cardoso A, Batista P. Displasia ectodérmica: relato de quatro casos com baixa expressividade. JBP J Bras Odontopediatr Odontol Bebê. 1998; 1:49-56. [ Links ]
6. Sarmento VA, Tavares RB, Vilas-Boas R, Ramalho LM, Falcão AF, Meyer GA. Displasia ectodérmica: revisão da literatura e relato de casos clínicos. Sitientibus. 2006; 34:87-100. [ Links ]
7. Sannomiya EK, Prado MC, Barrella B, Goldenberg FC. Displasia ectodérmica: aspectos clínicos e radiográficos. Assoc Bras Radiol Odontol. 2005; 6:12-6. [ Links ]
8. Muzio LO, Bucci P, Carife F. Prosthetic rehabilitation of a child affected from anhydrotic ectodermal dysplasia: a case report. J Contemp Dent Pract. 2005; 6:1-5. [ Links ]
9. Lind LK, Blicks CS, Lejon K. EDAR mutation in autosomal dominant hypohidrotic ectodermal dysplasia in two Swedish families. BMC Med Genet. 2006; 7:1-8. [ Links ]
10. Kramer FJ, Baethge C, Tschernitschek H. Implants in children with ectodermal dysplasia: a case report and literature review. Clin Oral Implants Res. 2007; 18:140-6. [ Links ]
11. Rad AS, Siadat H, Monzavi A, Mangoli A. Full mouth rehabilitation of a hypohidrotic ectodermal dysplasia patient with dental implants: a clinical report. J Prosthodont. 2007; 16:209-13. [ Links ]
12. Almeida SF, Solari HP. Displasia ectodérmica, ectrodactilia e fissura lábio-palatal: manifestações oculares da síndrome em relato de caso. Arq Bras Oftalmol. 2007; 70:125-8. [ Links ]
13. Akhyani M, Kiavash K. Ectodermal dysplasia with alopecia, onychodysplasia, hypohidrosis, keratoderma, abnormal teeth and deafness. Indian J Dermatol Venereol Leprol. 2007; 73:409-11. [ Links ]
14. Fabel J, Filho JS, Leonardo MM, Pena GP. Apresentação clínica da displasia ectodérmica hipoidrótica no recém-nascido: a propósito de um caso. An Bras Dermatol. 1999; 74:375-8. [ Links ]
15. DiFazio MP, Levin S, Depper M. Ectodermal dysplasia and brain cystic changes: confirmation of a novel neurocutaneous syndrome. J Child Neurol. 2002; 17:475-8. [ Links ]
16. Rouse C, Siegfried E, Breer W, Nahass G. Hair and sweat glands in families with hypoidrotic ectodermal dysplasia. Arch Dermatol. 2004; 140:850-5. [ Links ]
17. Sasaki Y, Kaida C, Saitoh I, Fujiwara T, Nonaka K. Craniofacial growth and functional change in oligodontia with ectodermal dysplasia: a case report. J Oral Rehabil. 2007; 34:228-35. [ Links ]
18. Yared FN, Oliveira VM, Lopes AC. Diagnóstico em Odontopediatria: caso atípico de displasia ectodérmica do tipo autossômico dominante. JBP J Bras Odontopediatr Odontol Bebê. 2000; 3:9-14. [ Links ]
19. Wright JT, Grange DK, Richter MK. Hypoidrotic ectodermal dysplasia. In: Pagon RA, Bird TD, Dolan CR, Stephens K. GeneReviews; [Internet]. [cited 2009 Jul 15]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1112 [ Links ]
20. Giust AC, Pellison F, Mendes WS, Melo LL. Criança com displasia ectodérmica: diagnósticos e intervenções de enfermagem baseados em NANDA e NIC. Arq Ciênc Saúde. 2006; 13:39-43. [ Links ]
21. Lexner MO, Bardow A, Bjorn-Jorgensen J, Hertz JM, Almer L, Kreiborg S. Anthropometric and cephalometric measurements in X-linked hypohidrotic ectodermal dysplasia. Orthod Craniofac Res. 2007; 10:203–15.
22. Resende L, Oliveira K, Glória J, Cruz R, Araújo C. Avaliação genética de paciente com displasia ectodérmica hipo-hidrótica. revisão de literatura e apresentação de caso clínico. Rev CROMG. 2003;
9:84-8.
23. Umérez C, Sosa RD, Simosa V. Síndrome de ectrodactilia - displasia ectodérmica - hendidura (EEC): revisión de la literatura; reporte de un caso. Acta Odontol Venez. 2002; 40:172-6. [ Links ]
24. Bal E, Baala L, Cluzeau C, El Kerch F, Ouldim K, Hadj-Rabia S, et al. Autosomal dominant anhidrotic ectodermal dysplasias at the EDARADD locus. Hum Mutat. 2007; 28: 703-9. [ Links ]
25. Xavier AS, Pelli PG, Benfatti SV, Bausells J. Displasia ectodérmica/anidrótica hereditária: revisão de literatura e relato de um caso clínico. Rev Ciênc Odontol. 2002; 5:81-5Links ] Arial, Helvetica, sans-serif">.
 Autor correspondente:
Autor correspondente:
Rúbia Alves Marques Hissa Ferreira
Av. Amazonas, 314/sl 1708 – Centro
CEP: 30180-001 – Belo Horizonte, MG, Brasil
E-Mail: rubiahissa@gmail.com
Recebido em 19/10/2010 – Aceito em 03/02/2011