








Serviços Personalizados
Artigo
 pdf em Inglês
pdf em Inglês Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailLinks relacionados
Compartilhar

 Permalink
PermalinkRevista Odonto Ciência (Online)
versão On-line ISSN 1980-6523
Rev. odonto ciênc. (Online) vol.26 no.1 Porto Alegre Jan. 2011
CASE REPORT
Apert syndrome: clinical and radiographic features and case report
Síndrome de Apert: características clínicas e radiográficas e relato de caso
Felipe Paes VaroliI; Karina Cecília Panelli SantosII; Claudio CostaII; Jefferson Xavier OliveiraII
IPaulista University, São Paulo, SP, Brazil
IIFaculty of Odontology, University of São Paulo, São Paulo, SP, Brazil
ABSTRACT
PURPOSE: Apert syndrome is a rare type I acrocephalosyndactyly syndrome characterized by craniosynostosis, severe syndactyly of the hands and feet, and dysmorphic facial features. Presents autosomal dominant inheritance assigned to mutations in the fibroblast growth factor receptors gene. The oral cavity of Apert patients includes a reduction in the size of the maxilla, tooth crowding, anterior open-bite of the maxilla, impacted teeth, delayed eruption, ectopic eruption, supernumerary teeth, and thick gingiva. The mandible usually is within normal size and shape, and simulates a pseudoprognathism.
CASE DESCRIPTION: A female patient, 13 years old, with diagnosis of Apert syndrome, attended a dental radiology clinic. The clinical signs were occular anomalies, dysmorphic facial features, syndactyly and oral features observed clinically and radiographically. The patient was referred to a specialized center of clinical care for patients with special needs.
CONCLUSION: Because of the multiple alterations in patients with Apert syndrome, a multidisciplinary approach, including dentists and neurosurgeons, plastic surgeons, ophthalmologists and geneticists, is essential for a successful planning and treatment.
Key words: Apert syndrome; acrocephalosyndactylia; craniosynostosis
RESUMO
OBJETIVO: A síndrome de Apert é um tipo raro de acrocefalossindactilia do tipo I caracterizada por cranioestenose, sindactilia severa das mãos e dos pés, e características faciais dismórficas. Apresenta herança autossômica dominante atribuída a mutações no gene referente aos receptores do fator de crescimento de fibroblastos. A cavidade oral de pacientes de Apert apresenta uma redução no tamanho da maxila, apinhamento dentário, mordida aberta anterior da mandíbula superior, dentes retidos, erupção atrasada, erupção ectópica, dentes supranumerários, gengival e espessura. A mandíbula é no tamanho e forma normais, e simula um pseudoprognatismo. Um caso de síndrome de Apert é apresentado.
DESCRIÇÃO DO CASO: Paciente do sexo feminino, 13 anos de idade, com diagnóstico de síndrome de Apert, foi atendida em um serviço de radiologia odontológica. A paciente apresentava anomalias oculares, características faciais dismórficas e sindactilia, aos exames clínico e radiográfico. A paciente foi encaminhada a um centro especializado para atenção a pessoas com necessidades especiais.
CONCLUSÃO: Uma abordagem multidisciplinar, incluindo cirurgiões-dentistas e neurocirurgiões, cirurgiões plásticos, oftalmologistas e geneticistas, é essencial para um planejamento bem sucedido e tratamento de casos de síndrome de Apert devido às múltiplas alterações nesses pacientes.
Palavras-chave: Síndrome de Apert; acrocefalossindactilia; craniosinostose
Introduction
Apert syndrome is a rare type I acrocephalosyndactyly syndrome characterized by craniosynostosis, severe syndactyly of the hands and feet, and dysmorphic facial features (1-3) that was first described by Apert, a French physician, in 1906 (1). Apert syndrome presents autosomal dominant inheritance assigned to mutations in the fibroblast growth factor receptors (FGFR-2) gene at locus 10q26 (2,4). Fibroblast growth factor receptors have a high affinity for fibroblast growth factors that, when bound to their specific receptors, play a role in signaling pathways with multiple biologic effects including cranial development and growth (3,5). In most cases the disorder results from a mutation in the father; its prevalence at birth is 1:65,0003 (2,6), and males and females may be affected with equal severity (3,5).
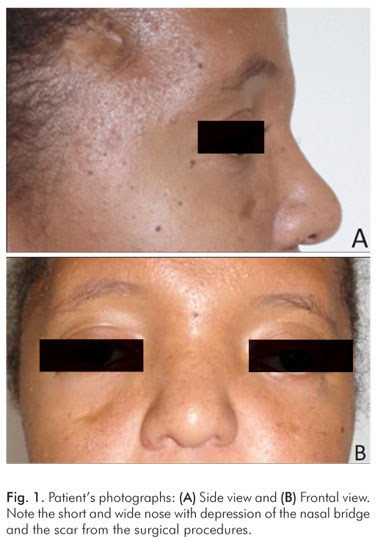
The clinical features are distinctive. The coronal suture fuses prematurely (at less than 3 months), leading to an acrocephalic (cone-shaped) head with shortened antero-posterior diameter, and a high prominent forehead. The midface is hypoplastic. Occular anomalies include hypertelorism, proptosis, and down slanting palpebral fissures. The nose is short and wide with depression of the nasal bridge (1,2,5,7). Previous studies report affected individuals with anomalies of the viscera, elbows and shoulders, skeleton and central nervous system, which often results in impaired mental function (5,6).
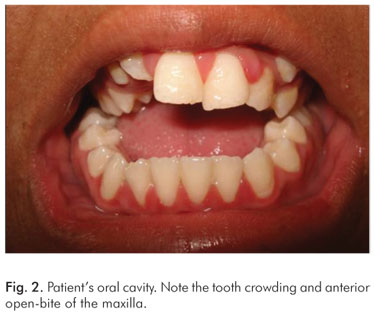
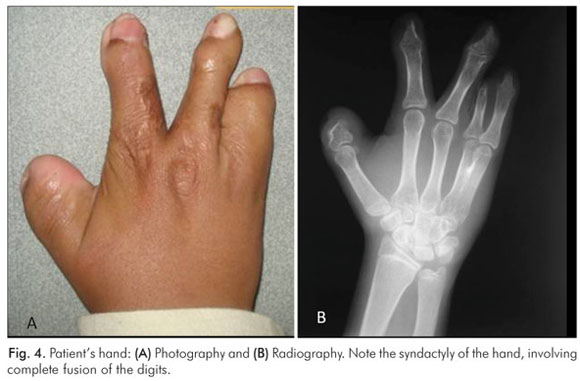
The oral cavity of Apert patients is also characteristic. The clinical findings include a reduction in the size of the maxilla, particularly in the antero- posterior direction. This reduction may result in tooth crowding and an anterior open-bite of the maxilla. The mandible is within normal size and shape, and simulates a pseudoprognathism. Dental anomalies such as impacted teeth, delayed eruption, ectopic eruption, supernumerary teeth, and thick gingiva are also common (3,5,7,8). Abnormalities of the upper and lower respiratory tracts include cleft soft palate, bifid uvula, Byzantine-arch palate, choanal stenosis, and anomalies of the tracheal cartilage (1). Mouth breathing, observed in most cases of Apert's syndrome, is related to alteration in facial growth (3). Syndactyly involving partial to complete fusion of the digits results in a spoon-like deformity of the hands and feet (1,2).
This article presents a case of Apert syndrome in a young female patient.
Case Description
Female patient, 13 years old, attended a radiology service for routine radiography exam requested by a general practitioner. The patient reported Apert syndrome and was the only case of her family. She had undergone two surgeries, at 4 months and 9 years of age, as recommended by a neurologist, to advance her forehead and face. The vision loss in the right eye was a sequelae of such surgical procedures.
The clinical features included occular anomalies (hypertelorism, proptosis, left eye divergent strabismus and down slanting palpebral fissures), short and wide nose with depression of the nasal bridge (Fig. 1). The oral cavity presented reduction in the size of the maxilla, tooth crowding and anterior open-bite of the maxilla (Fig. 2)
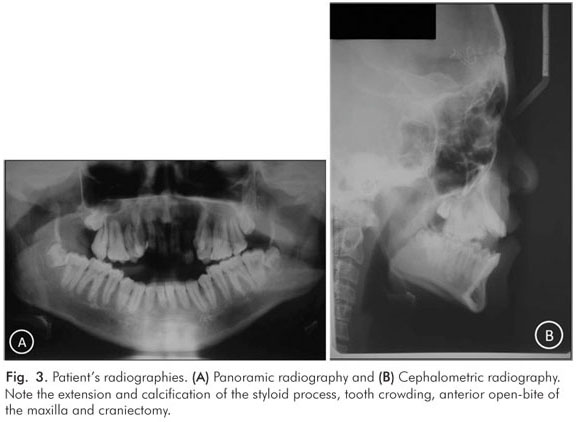
Radiographically, extension and calcification of styloid process, tooth crowding, anterior open-bite of the maxilla and cranioctomy were noted (Fig. 3). Syndactyly involving complete fusion of the digits of hands was present (Fig. 4). Abnormalities of the upper and lower respiratory tracts were not detected.
The patient was referred to a specialized center of clinical care for patients with special needs to receive a comprehensive healthcare treatment.
Discussion
Apert, in 1906, described the triad craniosynostosis, severe syndactyly of the hands and feet, and dysmorphic facial features, characterizing the syndrome (1-3). Later, a rare autosomal dominant heritage was linked to the syndrome, with mutations in the fibroblast growth factor receptors (FGFR-2) gene at locus 10q26 (2,4).
The clinical and oral features of Apert syndrome are well established and in agreement with the case described in the present study. The syndrome is clinically characterized by premature fusion of the coronal suture and hypoplastic midface (1,2,5,7). These are the main reasons for the previous surgical procedures mentioned by the patient. Occular anomalies, short nose with depression of the nasal bridge could also be observed. Syndactyly, as described by Apert (1,2), was also present. The oral cavity characteristics included reduction in the size of the maxilla, which may result in tooth crowding and an anterior open-bite of the maxilla (3,5,7,8) as seen in the patient. Effective clinical management to improve oral health usually requires the joint work of a periodontist and an orthodontist.
Some affected individuals have anomalies of the viscera, elbows and shoulders, skeleton and central nervous system (5,6) or abnormalities of the upper and lower respiratory tracts (1,3). However, the case reported here did not present any related complaint of these anomalies during clinical examination.
In conclusion, Apert syndrome is a rare autosomal heritage with many affected parts of the body. The integral healthcare delivery should include a multidisciplinary approach provided by dentists, neurosurgeons, plastic surgeons, ophthalmologists and geneticists for the effective planning and treatment of such patients.
References
1. DeGiovanni CV, Jong C, Woollons A. What syndrome is this? Apert syndrome. Pediatr Dermatol 2007;24:186-8. [ Links ]
2. Freiman A, Tessler O, Barankin B. Apert syndrome. Int J Dermatol 2006;45:1341-3. [ Links ]
3. Letra A, de Almeida AL, Kaizer R, Esper LA, Sgarbosa S, Granjeiro JM. Intraoral features of Apert's syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007;103:e38-41. [ Links ]
4. Dalben Gda S, das Neves LT, Gomide MR.Oral findings in patients with Apert syndrome. J Appl Oral Sci 2006;14:465-9. [ Links ]
5. Surman TL, Logan RM, Townsend GC, Anderson PJ. Oral features in Apert syndrome: a histological investigation. Orthod Craniofac Res 2010;13:61-7. [ Links ]
6. Carneiro GV, Farias JG, Santos FA, Lamberti PL Apert syndrome: review and report a case. Braz J Otorhinolaryngol 2008;74:640. [ Links ]
7. Albuquerque MAP, Cavalcanti MGP. Computed tomography assessment of Apert syndrome. Braz Oral Res 2004;18:35-9. [ Links ]
8. Verma S, Draznin M. Apert syndrome. Dermatol Online J 2005;11:15. [ Links ]
 Correspondence:
Correspondence:
Karina Cecília Panelli Santos
Disciplina de Radiologia – Faculdade de Odontologia da USP
Avenida Prof. Lineu Prestes, 2227 – Butantã
São Paulo, SP – Brasil
05508-000
E-mail: kapanelli@usp.br
Received: October 27, 2010
Accepted: February 2, 2011
Conflict of Interest Statement: The authors state that there are no financial and personal conflicts of interest that could have inappropriately influenced their work.